SeqCode: recoverChIPlevels
General description
Counting the number of ChIPseq/ATACseq/RNAseq reads on a set of genomic regions.
> bin/recoverChIPlevels -h SeqCode_v1.0 User commands recoverChIPLevels NAME recoverChIPLevels - a program to determine the ChIPseq/ATACseq/RNAseq avg/max/total counts of reads in a list of regions. SYNOPSIS recoverChIPLevels [-d][-l <bp>][-n][-s <reads>][-w <bp>][-v][-x prefix][-h] <chrom_info> <SAM file> <peaks.bed> <name> OUTPUT One folder with 1 file: - List of peaks + counts of ChIPseq/ATACseq/RNAseq reads. OPTIONS -d : Demo mode for small BAM files (min number reads control off). -l : Avg. fragment size (default: 150). -n : Normalize using the total number of reads. -s : Number of spike-in reads for reference-adjusted normalization (with -n). -w : Window resolution (default: 10). -v : Verbose. Display info messages. -x : Prefix for the output folder. -h : Show this help. SEE ALSO SeqCode homepage: http://ldicrocelab.crg.es GitHub source code: https://github.com/eblancoga/seqcode AUTHORS Written by Enrique Blanco. SeqCode_v1.0 User commands recoverChIPLevels
Examples
Example 1. Determining the ChIPseq levels of H3K4me3 in all the set of RefSeq transcripts in mouse.
(A) Counting the reads from the ChIPseq experiment on a set of genomic regions (gene bodies from RefSeq transcripts):
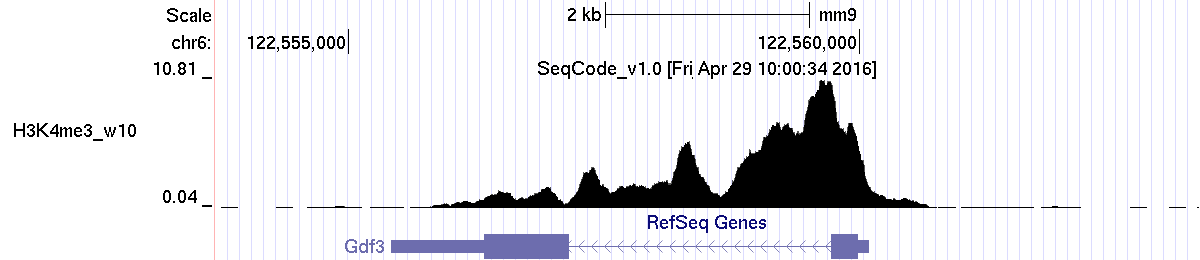
Example 1. The Gdf3 gene:
Example 1. The Nanog gene:
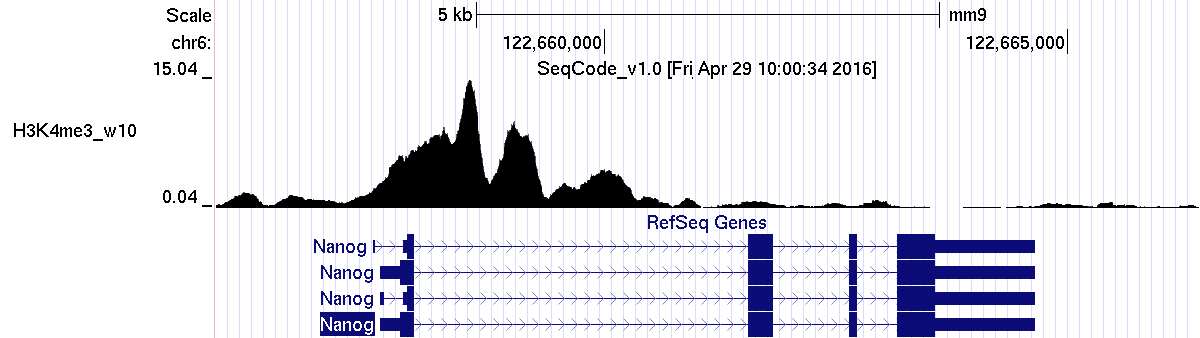
(A) Counting the reads from the ChIPseq experiment on a set of genomic regions (gene bodies from RefSeq transcripts):
> bin/recoverChIPLevels -vn ChromInfo.txt SRR1015741.bam refGene_transcripts.bed H3K4me3(B) Generating the profile for visualization in the UCSC browser (for visual confirmation):
> bin/buildChIPprofile -v -w 10 ChromInfo.txt SRR1015741.bam H3K4me3_w10
Example 1. The Gdf3 gene:
#chr pos1 pos2 locus average maximum total chr6 122555420 122560089 Gdf3-NM_008108 2.86 10.81 1331.70 (Max peak is 10.81)
Example 1. The Nanog gene:
#chr pos1 pos2 locus average maximum total chr6 122657506 122664651 Nanog-NM_001289831 2.40 15.04 1717.44 (Max peak is 15.04) chr6 122657582 122664651 Nanog-NM_028016 2.40 15.04 1698.48 chr6 122657582 122664651 Nanog-NM_001289830 2.40 15.04 1698.48 chr6 122657582 122664651 Nanog-NM_001289828 2.40 15.04 1698.48
ChIPseq and RNAseq samples
Please, follow the link below for further information on how to get and preprocess the raw data of
published ChIPseq and RNAseq samples utilized in this glossary of SeqCode functions.
[SAMPLES]
[SAMPLES]