SeqCode: processmacs
General description
Processing and cleaning the BedGraphs generated by the MACS software.
> bin/processmacs -h SeqCode_v1.0 User commands processmacs NAME processmacs - a program to clean MACS BedGraph profiles. SYNOPSIS processmacs [-c][-v][-h] <chrom_info> <bedgraph.gzip> <output name> <track name> OUTPUT BedGraph processed profile. OPTIONS -c : Compact averaged BedGraph profile. -v : Verbose. Display info messages. -h : Show this help. SEE ALSO SeqCode homepage: http://ldicrocelab.crg.es GitHub source code: https://github.com/eblancoga/seqcode AUTHORS Written by Enrique Blanco. SeqCode_v1.0 User commands processmacs
Examples
Example 1. Working with the output of MACS in the UCSC genome browser.
Before, let us assume that we have run MACS to find the peaks of one sample, using a second sample as a control:
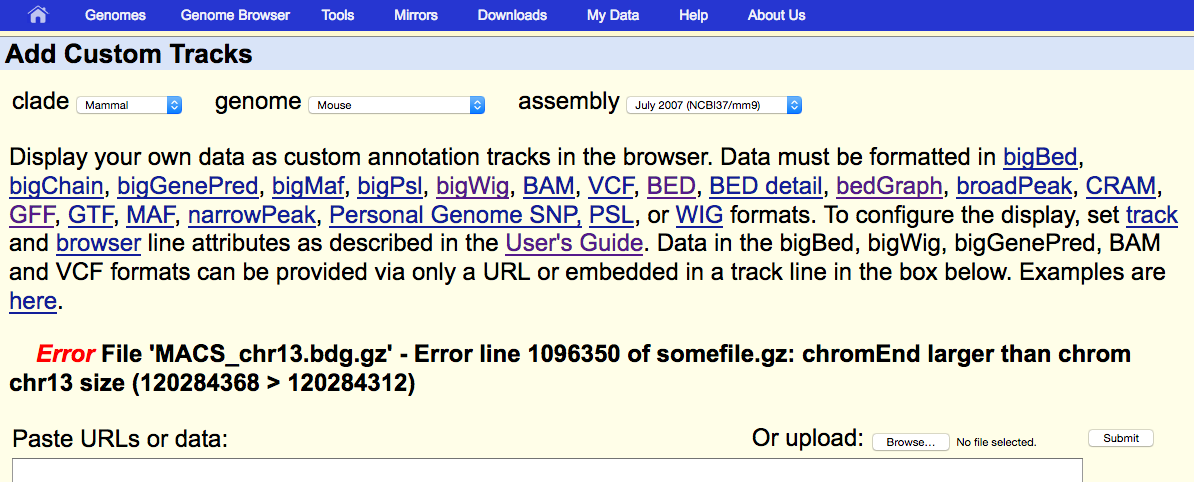
However, when we upload this profile in UCSC (mm9), we realize that something is wrong:
We run SeqCode for processing and cleaning this BedGraph file in mm9:

Example 2. Generating compact profiles that require a lower amount of space.
Now, we run SeqCode for processing and cleaning this BedGraph file in mm9 using the option -c:

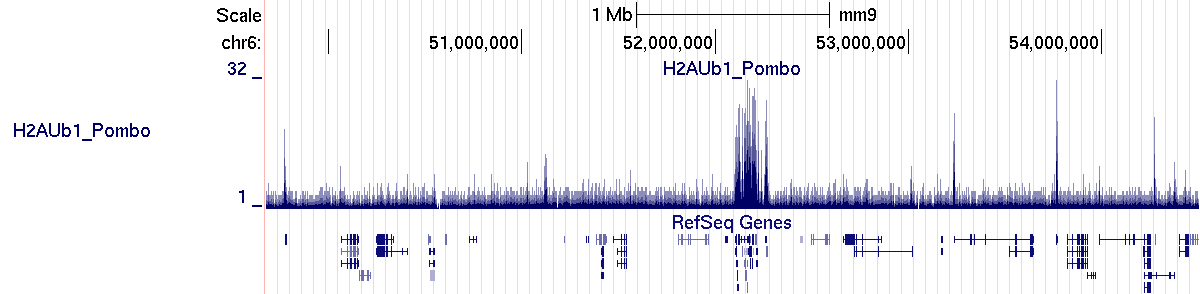
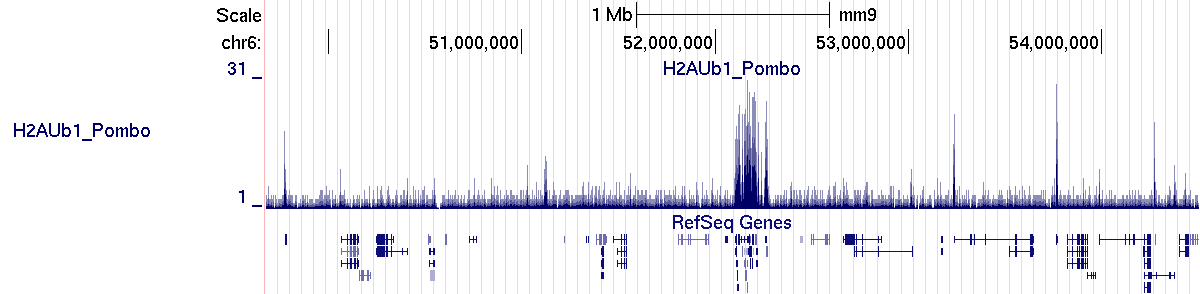
The profile generated by SeqCode is correctly uploaded into the UCSC genome browser interface:
Before, let us assume that we have run MACS to find the peaks of one sample, using a second sample as a control:
> macs14 -t H2AUb1_Pombo.bam -c ControlIP_Pombo.bam -g mm -n H2AUb1_Pombo -B -S --nomodel --shiftsize 75MACS, after running the previous command, has generated the BedGraph profile of the ChIPseq experiment:
However, when we upload this profile in UCSC (mm9), we realize that something is wrong:
We run SeqCode for processing and cleaning this BedGraph file in mm9:
> bin/processmacs -v ChromInfo.txt H2AUb1_Pombo_treat_afterfiting_all.bdg.gz H2AUb1_Pombo_nocompact.bdg H2AUb1_PomboThe new profile generated by SeqCode is correctly uploaded into the UCSC genome browser interface:
Example 2. Generating compact profiles that require a lower amount of space.
Now, we run SeqCode for processing and cleaning this BedGraph file in mm9 using the option -c:
> bin/processmacs -cv ChromInfo.txt H2AUb1_Pombo_treat_afterfiting_all.bdg.gz H2AUb1_Pombo.bdg H2AUb1_PomboWhen comparing to the previous profile, we see the performance of the reduction operation in the file size:
The profile generated by SeqCode is correctly uploaded into the UCSC genome browser interface:
ChIPseq and RNAseq samples
Please, follow the link below for further information on how to get and preprocess the raw data of
published ChIPseq and RNAseq samples utilized in this glossary of SeqCode functions.
[SAMPLES]
[SAMPLES]